Most people have received messages which at some point or other state "I would like to deposit 20,000,000.00 dollars in your account....". This is a common email fraud attempt which has been named the "Nigerian Prince" scam since the earliest versions of it were sent by a supposedly such member of royalty. You could read more about it in wikipedia, and a google search will show hundreds of related hits.
The messages, although very different, have a similar pattern. In this bio-recipe we will try to construct the phylogenetic tree of these messages and hence reconstruct how likely it is that people have copied and modified the original message(s) and created new versions. If the authors had copied from a single source and then modified the messages to their own taste, this procedure would recreate the tree quite well. However, it is possible that messages have been merged or copied from several sources, obscuring the process. The latter would be equivalent to lateral transfer of genetic information. It is also possible that similar messages have been reinvented from scratch. In any case, the whole procedure is an interesting example that may serve as a pattern for dealing with text, for constructing phylogenetic trees from diverse sources, separating the members in clusters and on some special forms of alignment. The resulting tree is also interesting in itself. Applications outside molecular biology are also possible, e.g. the construction of a phylogenetic tree of some piece of software that underwent development at different places/times, would follow exactly the same steps.
Reading the data: email messages
The raw email messages were collected for some time and kept in a standard plain text format. You can download the file from biorecipes.com, read it and do all the processing in Darwin.
all := ReadRawFile( 'NigerianPrince/mbox' ): length(all);
1095228
To store and process the messages we will define a class called Message . If it receives a single argument, it assumes that it is a raw message and proceeds to parse it. It is always convenient to write a method that will print the instances of the class, and finally we run CompleteClass which will generate some standard methods automatically.
Message := proc( id:string, shortid:string, subject:string,
header:string, body:string )
if nargs=1 then
if id[1..5] <> 'From ' then
error(id,'message does not start with From') fi;
i0 := SearchString( '\n', id );
if i0 < 0 then error(id,'does not have a single line') fi;
id1 := id[1..i0];
i1 := SearchString( '\n\n', id );
if i1 < 0 then error(id,'message does not appear to have a body') fi;
header1 := id[1..i1];
i2 := CaseSearchString( '\nSubject: ', header1 );
if i2 < 0 then subject1 := ''
else i3 := SearchString( '\n', i2+2+header1 );
subject1 := header1[ i2+11 .. i2+i3+2 ];
while length(subject1) >= 4 and
uppercase(subject1[1..4]) = 'RE: ' do
subject1 := 4+subject1 od
fi;
body1 := id[i1+3..-1];
Message( id1, ShortId(id1), subject1, header1,
ConvertMessage(body1) )
elif nargs=5 then noeval(procname(args))
else error(args,'invalid message format') fi
end:
Message_print := proc( m:Message )
printf( '\n%s (%s)\nSubject: %s\n\nbody: %s\n',
m[id], m[shortid], m[subject], m[body] )
end:
CompleteClass(Message):
Converting the text to a comparable form
The following function converts a general text into a form suitable for comparison and alignment. The messages are written in an alphabet of 26 characters where upper and lower case letters as well as numbers and special symbols are used. The mapping of lowercase characters to uppercase characters, or the grouping of several symbols in the same class (e.g. punctuation) can be done with a mapping function included in the Dayhoff matrices.
A new method ConvertMessage does this conversion, character by character. We search each character of the message in a string which contains all letters and digits (case-independent search). This is a simple and efficient way to find out if our character is a letter or a digit. The boolean variable spacing is used to keep only one spacing character per group. We will keep the first one, the rest will be just ignored. Finally, the result is accumulated in r , which may be shorter than the original string. One final exception is that in no case underscore characters are allowed in sequence alignment. The reason for this is that underscores indicate gaps, so if they would be allowed as sequence characters it would not be possible to determine where deletions happen. We transform all underscores to dashes.
ConvertMessage := proc( s:string )
ls := length(s);
r := CreateString(ls);
lr := 0;
spacing := true;
for i to ls do
k := SearchString(s[i],'abcdefghijklmnopqrstuvwxyz0123456789');
if k >= 0 then
lr := lr+1; r[lr] := s[i]; spacing := false
elif not spacing then
lr := lr+1;
r[lr] := If( s[i]='_', '-', s[i] );
spacing := true
fi
od;
r[1..lr]
end:
At this point it is convenient to define the function that will map the characters to positions in the scoring matrix. In our terminology this is a Mapping function. Numbers are rare in these messages, and their actual value does not have a very relevant meaning, so we will group all digits in a single symbol. The dimension of the scoring matrix will be 28. This is composed of 26 letters, 1 symbol for any of the 10 digits and 1 symbol for spacing and punctuation. Using the convenience of SearchString this is written as:
Dim := 28:
map := proc( ch:string )
if length(ch) <> 1 then error('invalid argument') fi;
k := SearchString(ch,'abcdefghijklmnopqrstuvwxyz');
if k >= 0 then k+1
elif SearchString(ch,'0123456789') >= 0 then 27
else 28 fi
end:
The function ShortId is an auxiliary function to select the first part of the sending email address. This is typically the User-ID, and it is intended as a quick identification of the email message. It searches for an "@" or a blank as a separator and return whatever comes after the "From" until the separator.
ShortId := proc( s:string )
if s[1..5] = 'From ' then procname( 5+s )
else
i := SearchString( '@', s );
if i > 0 then return( s[1..i] ) fi;
i := SearchString( ' ', s );
if i > 0 then return( s[1..i] ) fi;
s
fi
end:
Now we are ready to process all the messages in the string "all". Each message starts with a new line with the word "From" followed by a space, which is the standard email convention. We place all the messages in a list.
msgs := []:
do i := CaseSearchString( '\nFrom ', all );
if i < 0 then
if length(all) > 10 and all[1..5] = 'From ' then
msgs := append( msgs, Message(all) ) fi;
break
else msgs := append( msgs, Message(all[1..i+1]) );
all := i+1+all
fi
od:
For curiosity and checking, we can print one message.
print(msgs[2]);
From petersigho@management.com Fri, 3 Nov 2000 21:27:40 (petersigho) Subject: INVESTMENT PROPOSAL body: ECO INTERNATIONAL BANK HEADQUARTERS PLOT 84 AJOSE ADEOGUN STREET VICTORIA ISLAND LAGOS ATTN:PRESIDENT/CHAIRMAN STRICTLY A PRIVATE BUSINESS PROPOSAL I am DR PETER IGHOa manager in the Bills and Exchange at the Foreign Remittance Department of the ECO INTERNATIONAL BANK I am writing this letter to ask for your support and cooperation to carry out this business opportunity in my department.We discovered an abandoned sum of$15,000,000.00 Fifteen million United States Dollars only)in an account that belongs to one of our foreign customers who died along with his entire family of a wife and two children in November 1997 in a Plane crash.Since we heard of his death,we have been expecting his next-of-kin to come over and put claims for his money as the heir,because we cannot release the fund from his account unless someone applies for claim as the next-of-kin to the deceased as indicated in our banking guidelines.Unfortunately,neither their family member nor distant relative has everappeared to claim the said fund.Upon this discovery,I and other officials in my department have agreed to make business with you and release the total amount into your account as the heir of the fund since no one came for it or discovered he maintained account with our bank,otherwise the fund will be returned to the banks treasury as unclaimed fund.We have agreed that our ratio of sharing will be as stated thus;20 for you as foreign partner,75 for us the officials in my department and 5 for the settlement of all local and foreign expences incurred by us and you during the course of this business.Upon the successful completion of this transfer,I and one of my colleagues will come to your country and mind our share.It is from our 75 we intend to import Agricultural Machineries into my country as a way of recycling the fund.To commence this transaction,we require you to immediately indicate your interest by a return e-mail and enclose your private contact telephone number,fax number full name and address and your designated bank coordinates to enable us file letter of claim to the appropriate departments for necessary approvals before the transfer can be made.Note also,this transaction must be kept STRICTLY CONFIDENTIAL because of its nature.I look forward to receiving your prompt response.FAX NUMBER234-1-7592911 Best Regarde DR PETER IGHO
Eliminating identical duplicates
Pairs of messages which are identical serve no purpose whatsoever. They will be part of the same subtree, they contribute no useful information about mutations (there are none) but cost a lot of additional computation. Hence it is desirable to eliminate the exact repetitions. The following loop compares every pair of messages and, if they are identical, removes the second one from the list. Since the list may shorten during the process, the loops are controlled by a while condition.
for i while i <= length(msgs) do
for j from i+1 while j <= length(msgs) do
if msgs[i,body] = msgs[j,body] then
printf( 'msg %s is a duplicate of\n %s; removed\n',
msgs[j,id], msgs[i,id] );
msgs := [ op(msgs[1..j-1]), op(msgs[j+1..-1]) ];
fi
od
od;
msg From peterjohn2004@go.com Wed Feb 26 12:10 MET 2003 is a duplicate of
From peterjohn@ny.com Wed Feb 26 10:43 MET 2003; removed
msg From benwills9@netzero.com Sat Apr 5 12:38 MET 2003 is a duplicate of
From benwills11@netzero.com Sat Apr 5 12:28 MET 2003; removed
As expected, the exact duplicates come from (nearly) the same sender and at about the same time. This is expected from spam.
Building the Dayhoff matrices for this particular data
Running the all-against-all of 348 messages requires too much time for a normal interactive session (60378 long alignments). We will set a flag, named PartialRun , under which we can run just a few entries to show the process while the complete results are read from a file. Conversely, if PartialRun is not set, the complete computation will be run, and the files of results will be stored.
PartialRun := true;
PartialRun := true
The Dayhoff matrices used for protein alignments are completely useless in this case. The symbols do not correspond to amino acids, and their mutation probabilities will certainly differ. This is a situation that may be encountered quite often in biology too when we are faced with sequences which are not average proteins. So the following analysis is also very relevant for biology - it is the standard procedure to derive Dayhoff matrices from new data.
The procedure for finding the similarity/scoring matrices is iterative. It repeats the following steps:
| (1) | (first time only) Select an initial Dayhoff matrix which is sufficiently diagonally dominant. That is, identical matches will be positive and mismatches will be negative. We start with a matrix at PAM 30 which has all mutations equally likely. We will call this matrix DM1. |
| (2) | Align all the sequences against each other with this similarity matrix. |
| (3) | Select a subset of the alignments which ensures that there is no relative over-representation of some evolutionary event. This can be achieved by using a circular tour of the sequences with accumulated maximal similarity. It can also be achieved with the methods described for the Unbiased selection of sample alignments |
| (4) | From each of the selected alignments, count the matches and mismatches per symbol pair. Mismatches are counted 1/2 for each pair, as we do not know which one mutated into which. |
| (5) | Compute the frequencies, mutation matrix and Dayhoff matrices from the Counts matrix. (Darwin's ComputeDayMatrices command.) |
| (6) | Repeat from step 2 with these new matrices until convergence. Convergence is normally very fast, and in this example we will run a single iteration. |
For the initial matrix, it is easier to use the darwin function CreateDayMatrices which requires a matrix of counts for each pair of symbols. We will produce a synthetic counts matrix with all 1's except for the diagonal (10's).
SynCounts := CreateArray(1..Dim,1..Dim,1): for i to Dim do SynCounts[i,i] := 10 od: CreateDayMatrices( SynCounts, map, type=Alphabetic ); DM1 := SearchDayMatrix(30,DMS);
DM1 := DayMatrix(Alphabetic, pam=30.2968, Sim: max=13.157, min=-5.673, Mapping= map, dim=28, del=-26.627-1.396*(k-1))
Next we run the complete or restricted all-against-all matching:
n := If( PartialRun, 6, DB[TotEntries] );
AllAll := CreateArray(1..n,1..n):
Dist := CreateArray(1..n,1..n):
for i to n do
for j from i+1 to n do
AllAll[i,j] := AllAll[j,i] :=
Align(msgs[i,body],msgs[j,body],DM1);
Dist[i,j] := Dist[j,i] := "[Score];
od od:
maxdist := max(Dist);
n := 6 maxdist := 14539.1689
Find a circular tour of the leaves to collect mutation information
The set of all messages, assuming that they are all related, will form a phylogenetic tree. This tree is at this point unknown (and is what we want to construct). An excellent way to inspect all the pairs of leaves of the tree is by doing a circular tour of the tree in any of its planar representations. In the tour, we consider each pair of leaves and use the information of the path that connects both leaves. Each of the tree branches is considered exactly twice. This is very important; we do not want to over-represent information. Notice that in an all-against-all comparison, one would use some of the edges much more often than others.
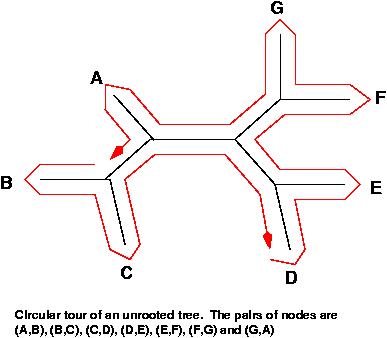
If shorter edges mean higher similarity, a planar tour has a minimum total sum of edges and a maximum total similarity. A circular tour with maximal similarity can be found by solving the Traveling Salesman Problem (TSP). The function ComputeTSP minimizes a tour, so we will complement the similarities such that by finding a minimum of the complements, we find the desired maximum. For convenience, we complement against the maximum so that all distances end up positive.
for i to n do for j from i+1 to n do
Dist[i,j] := Dist[j,i] := maxdist - Dist[i,j]
od od;
t := ComputeTSP( Dist );
t := [op(t),t[1]];
t := [1, 3, 5, 4, 2, 6] t := [1, 3, 5, 4, 2, 6, 1]
For programming convenience we have added the first sequence of the tour to the end. We are now ready to follow the tour and, for each alignment, count all the matches in a Count matrix.
Counts := CreateArray(1..Dim,1..Dim,If(PartialRun,1,0)):
if PartialRun then for i to Dim do Counts[i,i] := 10 od fi;
for i to n do
dps := DynProgStrings( AllAll[t[i],t[i+1]] );
for j to length(dps[2]) do
i1 := map(dps[2,j]);
i2 := map(dps[3,j]);
Counts[i1,i2] := Counts[i1,i2] + 1/2;
Counts[i2,i1] := Counts[i2,i1] + 1/2;
od;
od:
Next we compute the new Dayhoff matrices:
CreateDayMatrices(Counts,map,type=Alphabetical);
logPAM1 matrix contains negative off-diagonal entries This means that the count matrix may be too sparse or not representative of markovian evolution Try collecting larger samples from shorter distances
For the partial runs, we usually have too few sequences, which means that some counts are left empty. When there are empty counts, it becomes impossible to compute the scoring matrix. To prevent empty counts for the partial run, we start with a 1 on each count and a 10 on each diagonal.
Compute the all-against-all distance matrix
Now we are ready (by having a better scoring matrix) to align the messages and estimate their distances. This is done with the function Align when it receives an array of Dayhoff matrices instead of a single one (e.g. DMS). The function will compute the estimated distance and its variance for every pair of messages. The distance in this case corresponds to a Markovian model of random independent mutations (which is not exactly what happens to the messages, but works as an approximation). A final technical problem is caused by alignments which are spurious, very good and very short. This could happen by a very short phrase which appears in two unrelated messages. E.g. "thousand united states dollars". These short, good matches falsify the distance computations. To prevent this from happening, we use a mode of Align which forces an alignment of at least 400 characters of each message (parameter MinLength(400)). Have a look at the following code, but don't apply it right now, we will load a precomputed set of results in a minute:
for i to n do
for j from i+1 to n do
AllAll[i,j] := AllAll[j,i] :=
Align(msgs[i,body],msgs[j,body],DMS,MinLength(400));
od od:
bytes alloc=37.77Mb, time=125.970
If we were running a partial run, we would now read the full results from a pre-prepared file. We would also reset the number of messages and recompute the Dayhoff matrices. If this was a complete run, then we would create the file of results for future partial runs. For now, you can download the file 'AllAll.drw' here (AllAll.drw.gz). Place it in your working directory and load it with the ReadProgram method. The next lines of code are merely for reference; once you load the downloaded file, you don't need to run these lines anymore.
if PartialRun then
ReadProgram( 'NigerianPrince/AllAll.drw' );
else OpenWriting( 'NigerianPrince/AllAll.drw' );
printf( 'n := %d:\n', n );
printf( 'Counts := %A:\n', Counts );
printf( 'CreateDayMatrices(Counts,map,type=Alphabetical);\n' );
printf( 'AllAll := CreateArray(1..n,1..n);\n' );
printf( 'AllAll:=%A:\n', AllAll );
OpenWriting( terminal );
fi:
logPAM1 matrix contains negative off-diagonal entries This means that the count matrix may be too sparse or not representative of markovian evolution Try collecting larger samples from shorter distances bytes alloc=43.49Mb, time=192.860
It is interesting to see some alignments at different PAM distances. To make this easier, we write a function which selects the pair of messages which are at a distance as close as possible to a given one and then prints their alignment. This will allow us to test the limit of quality of the alignments.
PrintAlignmentAt := proc( d:positive )
best := AllAll[1,2];
for i to n do for j from i+1 to n do
if |best[PamDistance]-d| > |AllAll[i,j,PamDistance]-d| then
best := AllAll[i,j]
fi
od od;
print( best )
end:
We now print alignments close to 30, 50 and 70 to see where the boundary is:
PrintAlignmentAt( 30 );
lengths=427,456 simil=1025.1, PAM_dist=30.2968, identity=45.4% assist us_ to__________ transfer the sum of_________________ Twenty Million Fiv |||||||....||||......|..|||||||||||||||||||||..|..|...|...|..||...|||||||||||||| assistance to enable me transfer the sum of US$30,500,000.00 Thirty Million,Fiv e Hundred Thousand United St_ates Dollars 20,500,000)into ______his her_______ a ||||||||||||||||||||||||||||||||||||||||||..|...|...||||||....|..|...|........|| e Hundred Thousand United St ates Dollars___________)into your private/company a ccount.This fund________________ resulted from an over-invoiced bill from contra |||||||||..||||||....|.....|..|.|||||||..|..|.||.|....|........|....|....||||||| ccount.Th_e fund came about as a result____ of a_________________________ contra cts awarded by us unde___________r the budget allocation to___________ my Minist ||.|||||||||..|..|.||.........|..||...|......|.........|..|.|......|..|||||||||| ct_ awarded______ and executed for ___________________and on behalf of my Minist ry and this bill has been approved for payment by the concerned authorities.The |||...|....|....|...|....|........|...|.......|..|...|.........|...........||||| ry_________________________________________________________________________.The contract has since been execu___ted,commissioned and the_______________________ |||||||||.||||....|....|....|....|||.|..........|...|..||.......|..|...|.......| contract was s______________upposed to b_______________e awarded to two foreign contractor was _________________________________________________________________ |||||||||||..||..|...|....|..|..|...|...|...|..|...|.......|...|......|.......|. contractor___s to the tune of US$180,000,000.00 One hundred and Eighty Million U _______________________paid the actual co__st of____________ the contract.W .....|......|.......|....|.|||||......|||..|.||||...........||||||||||||||| nited States Dollars)But in the_______ course of negotiation,the contract w
PrintAlignmentAt( 50 );
lengths=531,576 simil=416.0, PAM_dist=50, identity=36.5% of Contract Award_______________ Committee with the Federal Ministry of Agricul |||||||||||||||||||...|..........|||||||||||....|||||.......|||||||||||||....... OF CONTRACT AWARD AND MONITORING COMMITTEE O__F THE________ MINISTRY OF _______ ture.By the virtue of our position and ___________________________the power best ....|..|...|......|..|.||........||||||.....|...........|..|....|...|.||||||.... _______________________URB______AN AND RURAL DEVELOPMENT.MY DUTY AS EMPOWER_____ owed on us by the_________ government,we carefully and deliberately over invoice ..|||..|..|||||||.........||||||||||||..|.........|...|........|.. .||..|....|.| __ED______ BY THEMAURITIUS GOVERNMENT IS ______________________TO PROV_______IDE d the v_______________________________alue of some contract_____________________ .|||||.....|.........|......|.........||..|..|....|.....|||.......|..|.....|...| _ THE BASIC AMENITIES,SOCIAL RECRATIONAL _______________ACTIVITIES IN URBAN AND __________s th_____________________at we a_________________________________ward .....|....||||..|........|........||.....|...|..|........|.....|............|.|| RURAL AREAS,THIS PROGRAMM INCLUDES ASSISTANCE TO DEPRIVED LOCAL COMMUNITIES AND . . . . (5 output lines skipped) . . . . _______________________________________________________________________________u ...........|....|....|........|..|....|....|....|..|.......|....|..........|...| FURTHERMORE FROM THIS PROJECTS WE HAVE BEEN ABLE TO SECURED SOME REASONABLE AMOU ne of_________ Twenty-Two Million five_ Hundred Thousand United State Dollars US | ||||.|.|..|.|||||||| ..|||||||||.|...|||||||||||||||||||.....||....|||||||||.. NT OF U.S.21.8(TWENTY ONE MILLION EIGHT HUNDRED THOUSAND U_____.S____.DOLLARS __ $22,500,000 00)Now the contracts have been fully executed and commission and pay |..|...|...|..|...|...|.||......|....|....|...|||...|....|...|...|||||||| . .... ________________________ON____________________LY)AS CO___________MISSION FROM __ ment are about to be made to all the contractors who have successfully executed .....||.|..||.|..|..|....|..|...|...|||||||||||||...|....|....||..||..|........| ____VARI___OU______________________S CONTRACTORS R____________ES__UL____________ their contracts,the_________ over invoic .....|.........|.......|....|||||||||||| ___________________TING FROM OVER INVOIC
PrintAlignmentAt( 70 );
lengths=434,594 simil=459.0, PAM_dist=70, identity=39.7% account will be compensated with twenty percent 20%of the total sum if you_____ |||||||||....|..|.... |.|...|....|.... ||| | ...|..||||||||.| .|.. .. |||||.... account ____________in a_____________ny part______ of the wor_ld,which you prov ___ will stand____________________________________________ as the beneficiary___ ...|||||| |...........|...|........|..|....|.....|..|...||||||||||||||||||||.. ide,will then facilitate the transfer of this money to you as the beneficiary ne __ of______________________ the fun______________d.Eighty_______________________ ..||||...|..|..|.....|.....|||||. |..|....|..|...|| . .|...|.......|...|..|..|. xt of kin of Mr.Barry Kelly.The money will be paid into your account for us to s _ percent___________ 80%for we_______________ the ______offic____ials involved.N ...|. .|..|.....|..||||||||.||...|..|...|...||||..|..|.|. | .|...|| |........|. hare in the ratio of 60%for me and 40%for you.There is no risk at all___________ ote that we have done our home work very well in our country,this transaction is ...|||..|.|| |..|....|...|...|.|||||. |.|....|..|...|.......|||||||||||||||||| ___ th____e pap______________erwork for_____________________ this transaction wi safe_____________________ and__________________________________ guaranteed ___1 . ..||....|..|...|........|||||..|........|..|...|......|.......||||||||||.|.... ll be done by the attorney and my position as the Branch Manager guarantees the 00%risk free____________________________.If you are interested please send down . |.. ..|........|..|....|...........|||||||||||||||||||||||||||||| | .|....| successful execution of this transaction.If you are interested,please rep_______ to us account information,so that we will immediately seek a____pproval o_______ ..|..|.......|...........|..|....|..|..| |||||||||||||.....||....|| || .......|. _______________________________________ly immediately ___via the private email a ____f the fund on your be__________half from the________________________________ ........|. . .||||||||.|......|.|.|||.|....||||.|.......|...|....|....|.......| ddress below.Upon your response,I shal_____l then provide you with more details ___ relevant ...|||||||||| and relevant
We can see that the boundary is fuzzy, but at 50 there are very large portions of text which have been duplicated whereas at 70, the relation is dubious. Consequently, we will choose 50 as the limiting distance for a trustworthy relation. It is interesting to notice that in Biology, we are used to consider and study homologies at much larger distances, up to 250 in a reliable way. Clearly the techniques that have been developed are much more sensitive than the human eye.
Now that we have computed all the distances, we will produce the full tree. We use the short IDs as a label with the concatenation of a sequential number to make sure that they are unique. Remember, you can display a tree in darwin by using the View method. If your linux machine does not have gv installed, you could use a method like this:
Eview := proc() CallSystem('evince temp.ps') end:
But now let's build some trees:
Labels := [ seq( msgs[i,shortid].i, i=1..length(msgs) )]: Seqs := [ seq( m[body], m=msgs )]: tree := PhylogeneticTree( Seqs, Labels, Distance, AllAll ): DrawTree( tree, Unrooted, CrossReference );
bytes alloc=57.98Mb, time=213.140 bytes alloc=62.18Mb, time=214.210 bytes alloc=62.18Mb, time=215.280 bytes alloc=62.18Mb, time=274.030
The full tree is quite large and impossible to read. To improve this we will do several things. First we will not print branch lengths nor nodes. In these cases, the Phylogram style is the most readable format. We will use the reordering that makes the left side the largest one so that the top of the tree appears more organized. Because the tree is so large, we will trim it down by removing the entries which are 80% or more identical. To do this, we will use the fact that we have constructed a tree based on distances; hence, very similar sequences will have a very short distance and will be neighbour leaves in the tree. Using this idea, we write a function that returns a trimmed tree:
TrimTree := proc( t:Tree, allall:matrix )
if type(t,Leaf) then
t;
else
l := TrimTree( t[Left], allall );
r := TrimTree( t[Right], allall );
if type(l,Leaf) and type(r,Leaf) and
allall[l[3],r[3],Identity] > 0.8 then
printf( '%s and %s have identity %.2f%%, %s ignored\n',
l[Label], r[Label], allall[l[3],r[3],Identity]*100,
r[Label] );
l;
else
Tree(l,t[2],r);
fi;
fi;
end:
With all these changes, the tree looks now like this:
DrawTree( TrimTree(tree,AllAll), Phylogram, Legend,
OrderLeaves=LeftHeavy, LengthFormat='' );
luckygarbi197 and simonmuzendda187 have identity 95.75%, simonmuzendda187 ignored nelsonbrown12009281 and nelsonbrown12013260 have identity 98.92%, nelsonbrown12013260 ignored charlesdorman222 and judeagada1199 have identity 91.71%, judeagada1199 ignored benwills20012002150 and benwills11146 have identity 96.84%, benwills11146 ignored jomomoh_200a149 and jomomoh77133 have identity 99.44%, jomomoh77133 ignored kudibrowne111320 and genkudi198 have identity 98.01%, genkudi198 ignored kubrown77a206 and kubrown77188 have identity 99.97%, kubrown77188 ignored kudibrowne111320 and kubrown77a206 have identity 96.80%, kubrown77a206 ignored charles2003321 and fred.peters309 have identity 96.45%, fred.peters309 ignored jameskyari1159 and jameskyari160 have identity 99.93%, jameskyari160 ignored ifekmark319 and markinjoha284 have identity 96.14%, markinjoha284 ignored hasan210k271 and mk_hassan002339 have identity 98.32%, mk_hassan002339 ignored o_attah204 and drbenakiga294 have identity 82.36%, drbenakiga294 ignored wallmark280 and petersigho2 have identity 94.81%, petersigho2 ignored challenge_1183 and challenge_sec179 have identity 97.72%, challenge_sec179 ignored johndisi1973254 and bmakelele1233 have identity 99.97%, bmakelele1233 ignored johndisi1973254 and nomvete12 have identity 86.52%, nomvete12 ignored Warning: DynProgStrings could not reach score 11406.79144, reached 11341.0020119 instead seyiademola209 and seyiademola200 have identity 99.95%, seyiademola200 ignored gamadasun55 and obasekig50 have identity 92.93%, obasekig50 ignored . . . . (29 output lines skipped) . . . . ladawamobutu3000166 and ladawa_mobutu147 have identity 99.96%, ladawa_mobutu147 ignored ladawamobutu3000174 and ladawamobutu3000166 have identity 99.67%, ladawamobutu3000166 ignored sesekom324 and ladawamobutu3000174 have identity 94.44%, ladawamobutu3000174 ignored seseko13 and sesekom324 have identity 87.50%, sesekom324 ignored sekmobut109 and seseko13 have identity 93.31%, seseko13 ignored kurumamarvin236 and kurumamarvin224 have identity 99.89%, kurumamarvin224 ignored solomongarba22235 and bulawa2000108 have identity 96.81%, bulawa2000108 ignored macheke33160 and vicikesa273 have identity 87.76%, vicikesa273 ignored simonmuzenda152 and simonmuzenda113 have identity 98.60%, simonmuzenda113 ignored sodindo200140 and sodindo200154 have identity 99.94%, sodindo200154 ignored mahabile50207 and mahabile10182 have identity 99.54%, mahabile10182 ignored danstevens101249 and dsmnd23245 have identity 85.45%, dsmnd23245 ignored donaldste10247 and jamesufon5050212 have identity 96.79%, jamesufon5050212 ignored donaldste10247 and emmastevens100226 have identity 99.02%, emmastevens100226 ignored rubutuwena99202 and rubutuwena99201 have identity 99.72%, rubutuwena99201 ignored franksalihu2320 and musa10smith336 have identity 94.01%, musa10smith336 ignored rubutuwena99202 and franksalihu2320 have identity 95.29%, franksalihu2320 ignored donaldste10247 and rubutuwena99202 have identity 91.57%, rubutuwena99202 ignored donaldste10247 and emmastevens100246 have identity 97.23%, emmastevens100246 ignored
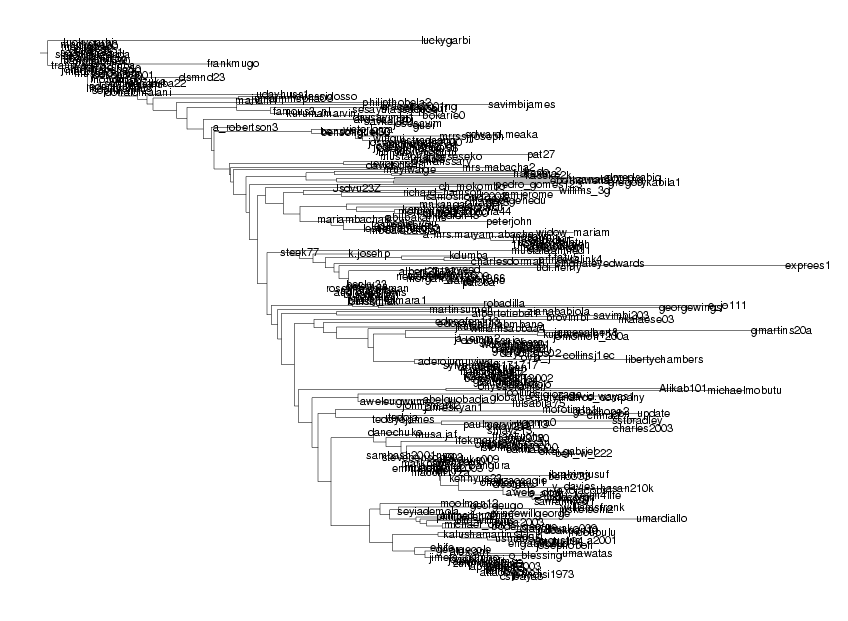
... which shows some of the structure, but still virtually impossible to find the details.
Separating the sequences into clusters
Sequences which are completely unrelated to one another disrupt the phylogenetic trees. Sometimes this disruption is so great that it is difficult to recognize the true properties. This can very well happen in our case where there may be more than a single source of messages. Like with genomic sequences, trying to associate completely different classes of objects destroys real relations. Once that we have computed the all-against-all alignments, we can cluster the sequences using the Clusters function in Darwin. This function provides a distance parameter to make the clusters; hence, the algorithm can be tuned to the data. In this case we know that we would like to separate the clusters around distance 50
clusters := Clusters( AllAll, AveDistance=50 ):
These clusters are composed of truly related messages. We write a function to compute a tree for a given cluster. This function receives a set of Entry numbers (a cluster) and uses the AllAll matrix to select the desired alignments; no further computation of alignments is needed. The rest is identical to the previous tree construction.
ClusterTree := proc( c:set(posint) )
n2 := length(c);
allall := CreateArray(1..n2, 1..n2);
for i to n2 do for j from i+1 to n2 do
allall[i,j] := allall[j,i] := AllAll[c[i],c[j]];
od od;
Labels := [ seq( msgs[i,shortid].i, i=c )];
Seqs := [ seq( msgs[i,body], i=c )];
tree := PhylogeneticTree( Seqs, Labels, Distance, allall );
DrawTree( TrimTree(tree,allall), Unrooted, CrossReference );
end:
We now test the function over three largest clusters. To do this, we first sort the clusters by reverse size (so the three largest are the first three) and display the two largest.
clusters := sort( clusters, x -> -length(x) ): clusters[1]; clusters[2];
{16,20,27,40,52,54,73,99,108,113,117,127,131,136,137,152,155,160,182,186,196,201
,202,207,212,221,226,235,246,247,256,269,276,287,295,318,323,336}
{5,26,28,36,105,107,141,179,181,205,213,219,220,227,241,259,273,306}
ClusterTree( clusters[1] );
simonmuzenda113 and muzendaasimon52 have identity 97.78%, muzendaasimon52 ignored simonmuzenda152 and simonmuzenda113 have identity 98.60%, simonmuzenda113 ignored mahabile10182 and mahabile50207 have identity 99.54%, mahabile50207 ignored rubutuwena99201 and rubutuwena99202 have identity 99.72%, rubutuwena99202 ignored lindaibrahim3117 and sophikan12295 have identity 89.78%, sophikan12295 ignored transvageazimba287 and mbowenilizzy99 have identity 95.24%, mbowenilizzy99 ignored sodindo200140 and sodindo200154 have identity 99.94%, sodindo200154 ignored sadu25131 and sadu25318 have identity 96.35%, sadu25318 ignored sadu25131 and vicikesa273 have identity 97.92%, vicikesa273 ignored bulawa2000108 and solomongarba22235 have identity 96.81%, solomongarba22235 ignored
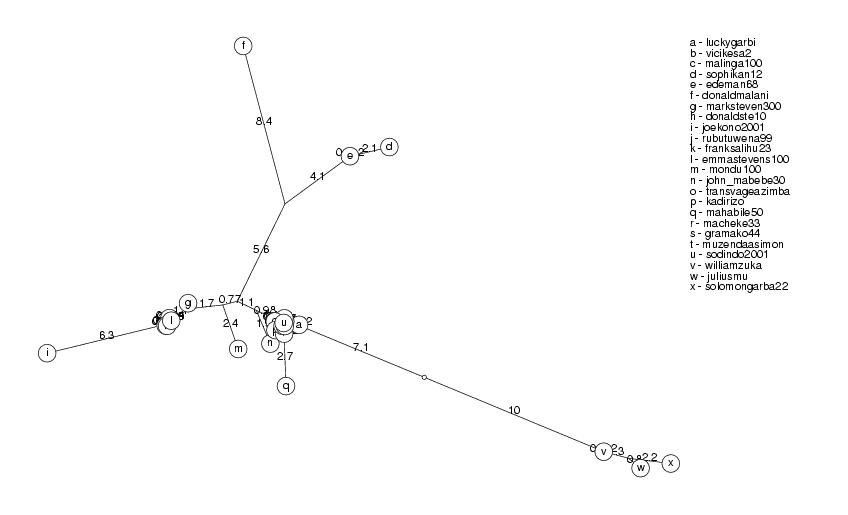
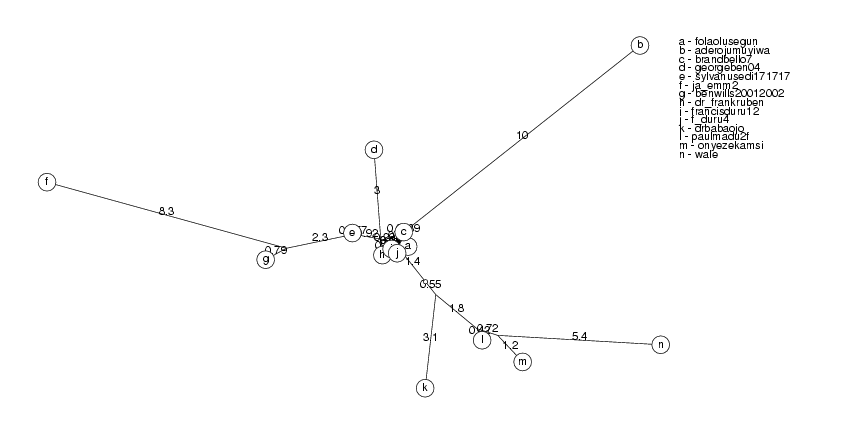
ClusterTree( clusters[2] );
pat2726 and colzaqb5 have identity 98.85%, colzaqb5 ignored josephmobutu56259 and jmobutu48213 have identity 98.76%, jmobutu48213 ignored bensonguei30227 and bensonguei30241 have identity 99.39%, bensonguei30241 ignored
ClusterTree( clusters[3] );
coleman348 and becky33267 have identity 96.73%, becky33267 ignored roseline.coleman90 and coleman348 have identity 95.85%, coleman348 ignored saratadobe216 and roseline.coleman90 have identity 93.82%, roseline.coleman90 ignored Warning: DynProgStrings could not reach score 33344.0193, reached 33265.5380248 instead Warning: DynProgStrings could not reach score 33344.0193, reached 33265.5380248 instead audreywilliams138 and saratadobe216 have identity 88.74%, saratadobe216 ignored Warning: DynProgStrings could not reach score 30141.8336, reached 30065.515849 instead nelsonbrown12013260 and nelsonbrown12009281 have identity 98.92%, nelsonbrown12009281 ignored nelsonbrown12013260 and nelsonbrown659 have identity 96.28%, nelsonbrown659 ignored
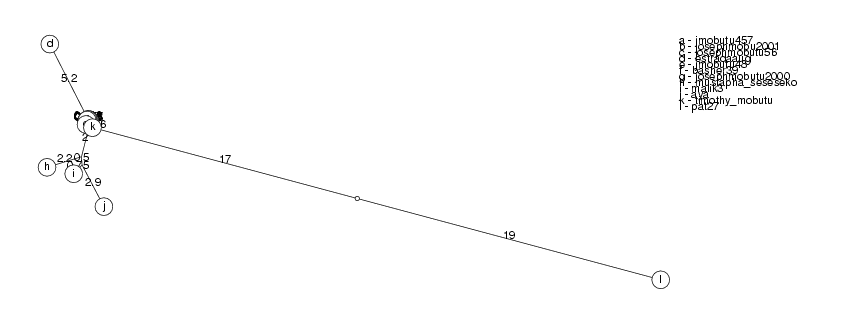
The clusters show a pattern which is typical of sequences which are mutated again and again and we have all the intermediate steps of their evolution (which is not the case in biology). This is indicated by nodes which have branches so short that they lie against the main branch. As we collect more information, it is likely that we will connect some of the disjoint clusters.
The source data for this bio-recipe (as an electronic mailbox standard format) can be found here.
© 2011 by Gaston Gonnet, Informatik, ETH Zurich
Please cite as:
@techreport{ Gonnet-NigerianPrince,
author = {Gaston H. Gonnet},
title = {Phylogenetic tree of the "Nigerian Prince" email scam},
month = { July },
year = {2003},
number = { 415 },
howpublished = { Electronic publication },
copyright = {code under the GNU General Public License},
institution = {Informatik, ETH, Zurich},
URL = { http://www.biorecipes.com/NigerianPrince/code.html }
}
Last updated on Thu Nov 3 13:25:53 2011 by zo
!!! This document is stored in the ETH Web archive and is no longer maintained !!!